Болезнь Вильсона-Коновалова
Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация) — это генетическое расстройство, не позволяющая организму удалять лишнюю медь, вызывая накопление меди в печени, головном мозге, глазах и других органах.
Для поддержания здоровья организму требуется небольшое количество меди из пищи, но большое количество меди вредно. Без лечения болезнь Вильсона может привести к высокому содержанию меди, что может привести к опасному для жизни повреждению органов.
Насколько распространена болезнь?
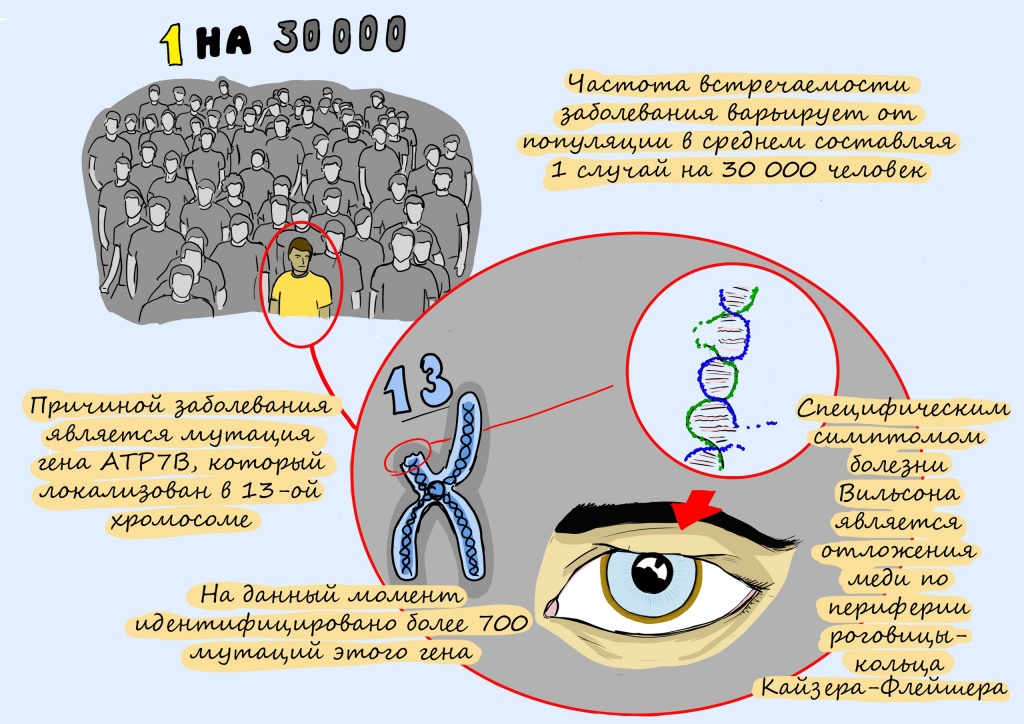
Эксперты все еще изучают, насколько распространена болезнь Вильсона. Более старые исследования показали, что больны примерно 1 из 30 000 человек. Эти исследования были проведены до того, как ученные обнаружили мутации генов, вызывающих заболевание.
Более новые исследования генов пациентов предполагают, что заболевание может быть более распространенной. Исследование, проведенное в Великобритании, показало, что примерно у 1 из 7000 человек есть генетические мутации, вызывающие болезнь Вильсона.
Эксперты не уверены, почему генные исследования предполагают, что заболевание встречается чаще, чем считалось ранее. Одной из причин может быть то, что некоторым больным пациентом не ставили диагнозы. Другая причина может заключаться в том, что некоторые пациенты имеют мутации генов, но заболевание по какой-то причине не развивается.
Причины болезни Вильсона-Коновалова
Как известно, болезнь Вильсона вызывают мутации гена под названием ATP7B. Эти мутации генов не позволяют организму удалить лишнюю медь из организма. Обычно печень выделяет лишнюю медь в желчь. Медь вместе с другими токсичными веществами и отходами выводится из организма через желудочно-кишечный тракт. При заболевании печень выделяет меньше меди в желчь, соответственно, в организме её остается больше.
Мутации ATP7B, вызывающие заболевание, наследуются, т.е., передаются от родителей ребенку. Мутация является аутосомно-рецессивной, что означает, что ребенок должен унаследовать две мутировавшие копии гена ATP7B, по одному от каждого родителя, чтобы заболеть. Люди, имеющие один ген ATP7B без мутации и один ген ATP7B с мутацией, обычно не страдают заболеванием, а являются её носителями.
Людям может быть присуща болезнь, если оба родителя являются носителями, не страдающими от неё сами. На приведенной ниже диаграмме показана вероятность наследования заболевания от родителей, которые являются носителями.

Мутации, вызывающие болезнь Вильсона, являются аутосомно-рецессивными, что означает, что человек должен унаследовать две копии гена с мутациями, чтобы заболеть.
Симптомы болезни Вильсона
Симптомы болезни Вильсона многообразны. Состояние присутствует при рождении, но симптомы не проявятся, пока не накопиться слишком много меди в печени, головном мозге или других органах.
Некоторые пациенты не испытывают признаков и симптомов гепатоцеребральной дистрофии до постановления диагноза и лечения. Однако, если симптомы присутствуют, они связаны с печенью, нервной системой и психическим здоровьем, глазами или другими органами.
Печеночные симптомы

У людей с болезнью Вильсона могут развиться признаки гепатита или воспаление печени. В некоторых случаях эти симптомы у больных появляются, когда возникает острая печеночная недостаточность.Симптомы включают:
- чувство усталости;
- тошнота и рвота;
- отсутствие аппетита;
- боль над печенью, в верхней части живота;
- потемнение цвета мочи;
- светловатый цвет стула;
- желтоватый оттенок склер глаз и кожи (желтуха)
Некоторые люди с заболеванием имеют симптомы только в том случае, если у них развивается хроническая болезнь печени и осложнения, вызванные циррозом печени. Эти симптомы могут включать:
- чувство усталости или слабости;
- беспричинное похудение;
- вздутие живота от скопления жидкости в животе (асцит);
- отек голеней, лодыжек или ступней;
- зуд кожи;
- желтуху.
Признаки со стороны нервной системы и психического здоровья
У пациентов с заболеванием могут развиться проблемы с нервной системой и психическим здоровьем. Эти проблемы чаще встречаются у взрослых, но иногда появляются и у детей. Симптомы нервной системы могут включать:
- проблемы с речью, глотанием или физической координацией;
- скованность мышц;
- тремор или неконтролируемые движения.
Симптомы психического здоровья могут включать:
- тревогу;
- изменения в настроении, личности или поведении;
- депрессию;
- психоз.
Глазные симптомы

Многие люди с болезнью Вильсона имеют кольца Кайзера-Флейшера, представляющие собой кольца зеленоватого, золотого или коричневатого цвета по краям роговицы. Кольца появляется в результате накопление меди в глазах. Так просто их не заметить, обычно врач-офтальмолог обнаруживает кольца во время специального обследования глаз, называемого осмотром щелевой лампой.
Среди пациентов с проблемами нервной системы при болезни Вильсона-Коновалова более 9 из 10 имеют кольца Кайзера-Флейшера, а среди больных, испытывающих проблемы с печенью, только 5-6 из 10 имеют кольца Кайзера-Флейшера.
Другие симптомы и проблемы со здоровьем
Гепатоцеребральная дистрофия может повлиять и на другие части тела, вызывая симптомы или проблемы со здоровьем, в том числе:
- тип анемии, называемой гемолитическая анемия;
- проблемы с костями и суставами, например, артрит или остеопороз;
- проблемы с сердцем, например, кардиомиопатия;
- проблемы с почками (ацидоз почечных канальцев и нефролитиаз).
У больных с гепатоцеребральной дистрофией симптомы обычно появляются в возрасте от 5 до 40 лет. Однако у некоторых пациентов признаки проявляются в более молодом или старшем возрасте. Врачи обнаружили первые симптомы болезни Вильсона у детей в возрасте 9 месяцев и у взрослых старше 70 лет.
Диагностика

Болезнь диагностируют на основании медицинской и семейной истории, физического осмотра, осмотра глаз и анализов.
Врачи обычно используют анализ крови и 24-часовой анализ мочи для диагностики болезни Вильсона. Специалисты также проводят биопсию печени и визуализационные исследования.
Медицинская и семейная история.
Врач спросит о семье и личной истории болезни Вильсона и других состояниях, вызывающих симптомы.
Во время медицинского осмотра врач проверит признаки повреждения печени, например:
- изменения кожи;
- увеличение селезенки или печени;
- припухлость на животе;
- отек в нижней части ног, ступней или лодыжек;
- желтоватый цвет белых глаз.
Обследование глаз проводиться щелевой лампой. Во время обследования щелевой лампой врач использует специальный свет для поиска колец Кайзера-Флейшера в глазах.
Для анализа крови медицинский работник возьмет образец крови и отправит ее в лабораторию.
Специалист может заказать один или несколько анализов крови, включая анализы, проверяющие количество:
- церулоплазмина, белка, несущего медь в крови. Люди с болезнью Вильсона часто имеют низкий уровень церулоплазмина, но не всегда.
- меди. У пациентов с заболеванием уровень меди в крови может быть ниже нормы. Острая печеночная недостаточность из-за болезни Вильсона приводит к повышению уровня меди в крови.
- аланинатрансаминаза (АЛТ) и аспартататрансаминаза (АСТ). Больные с гепатоцеребральной дистрофией могут иметь ненормальные уровни АЛТ и АСТ.
- эритроцитов для поиска признаков анемии.
Врачи также могут заказать анализ крови на наличие мутаций гена, вызывающих болезнь, если другие медицинские анализы не помогут подтвердить или исключить диагноз заболевания.
24-часовой анализ мочи.
В течение 24 часов пациент должен будет собирать свою мочу в домашних условиях в специальный контейнер, предоставив ее потом медицинским работникам. Медицинский работник отправит мочу в лабораторию, которая проверит количество меди в моче. Уровень меди в моче у пациентов с болезнью Вильсона-Коновалова часто выше нормы.
Если результаты анализов крови и мочи не подтверждают или не исключают диагноз заболевания, врач назначит биопсию печени. Во время биопсии врач извлечет небольшие кусочки ткани из печени, а патологоанатом исследует ткань под микроскопом, чтобы найти особенности конкретных заболеваний печени, такие как болезнь Вильсона, и проверит наличии повреждений и цирроза печени. Кусочек ткани печени будет отправлен в лабораторию, которая проверит количество меди в ткани.
Визуализационные методы исследования.
При проблемах с нервной системой врачи могут заказать визуальные тесты для выявления признаков болезни Вильсона или других состояний мозга.
- Магнитно-резонансная томография (МРТ), которая использует радиоволны и магниты для получения детальных изображений органов и мягких тканей без использования рентгеновских лучей.
- Компьютерная томография (КТ), которая использует комбинацию рентгеновских лучей и компьютерных технологий для создания детальных изображений.
Лечение болезни Вильсона-Коновалова
Лечат заболевание при помощи:
- лекарств, удаляющих медь из организма, называемых хелатирующими агентами;
- цинка, препятствующего поглощению меди кишечником.
Во многих случаях лечение улучшает или предотвращает симптомы и повреждения органов. Врачи также могут порекомендовать изменить диету, чтобы избежать употребления продуктов с высоким содержанием меди.
Люди с болезнью Вильсона-Коновалова нуждаются в пожизненном лечении. Прекращение терапии может привести к острой печеночной недостаточности. Медицинские работники регулярно будут проводить анализы крови и мочи, чтобы проверять, как протекает лечение.
Хелатирующие агенты
Пеницилламин (Купрамин, Депен) и Триентин (Сиприн) являются двумя хелатирующими агентами, используемыми для лечения болезни Вильсона. Лекарства удаляют медь из организма.
Пеницилламин чаще вызывает побочные эффекты, чем триентин. Побочные эффекты пеницилламина могут включать лихорадку, сыпь, проблемы с почками или проблемы с костным мозгом. Пеницилламин также снижает активность витамина В6, поэтому врачи могут порекомендовать принимать добавку витамина В6 вместе с пеницилламином. В некоторых случаях, когда пациенты с симптомами нервной системы начинают принимать хелатирующие агенты, их симптомы усиливаются.
Когда начинается лечение, врачи постепенно увеличивают дозу хелатирующих агентов. Больные принимают более высокие дозы хелатирующих агентов, пока лишняя медь из организма не будет удалена. Когда симптомы болезни Вильсона улучшатся, и анализы покажут, что содержание меди на безопасном уровне, могут назначить более низкие дозы хелатирующих агентов в качестве поддерживающей терапии. Препараты этой группы применяются с перерывами в течение всей жизни.
Хелатирующие агенты могут мешать заживлению ран, поэтому, на момент операции врачи могут назначать более низкие дозы препарата.
Цинка предотвращает поглощение меди кишечником. Врачи могут назначить цинк в качестве поддерживающей терапии, после того, как хелатообразующие агенты удалят лишнюю медь из организма. Также цинк может назначаться людям, у которых болезнь Вильсона-Коновалова обнаружена, но протекает бессимптомно.
Наиболее распространенным побочным эффектом цинка является расстройство желудка.
Прекращение приема лекарств от болезни может вызвать быстрое накопление меди и опасные для жизни последствия. Важно, чтобы пациенты, принимающие ацетат цинка, использовали рецептурную версию этого препарата, потому что пищевые добавки не могут быть биоэквивалентными и могут быть неэффективными.
Питание

Во время лечения, врач может порекомендовать избегать продуктов с высоким содержанием меди, например:
- шоколад;
- печень;
- грибы;
- орешки;
- моллюск.
После того, как лечение понизило уровень меди и началась поддерживающая терапия, поговорите с врачом о том, можно ли безопасно есть умеренное количество этих продуктов.
Если водопроводная вода поступает из колодца или течет по медным трубам, проверьте уровень меди в воде. Вода, проходящая в медных трубах, может содержать медь. Возможно, придется использовать фильтры для воды, чтобы удалить медь из водопроводной воды.
Из соображений безопасности поговорите с врачом перед использованием пищевых добавок, витаминов, или любых дополнительных или альтернативных лекарств. Некоторые пищевые добавки могут содержать медь.
Как лечат болезнь Вильсона беременным женщинам?
Беременные женщины должны продолжать лечение болезни на протяжении всей беременности. Врачи назначат более низкие дозы хелатирующих препаратов. Поскольку плод нуждается в небольшом количестве меди, снижение дозы может удерживать медь на безопасном уровне без удаления слишком большого количества.
В большинстве случаев врачи рекомендуют женщинам продолжать принимать полную дозу цинка во время беременности. Эксперты рекомендуют женщинам с болезнью Вильсона не кормить грудью, если принимают хелатирующие препараты. Пеницилламин присутствует в грудном молоке и может быть вредным для ребенка. О безопасности триентина и цинка, содержащегося в грудном молоке имеется мало информации.
Как лечат осложнения болезни?
Если заболевание приводит к циррозу, врачи лечат проблему и осложнения, связанные с циррозом, с помощью лекарств, операции и других медицинских процедур.
Если болезнь вызывает острую печеночную недостаточность или печеночную недостаточность вследствие цирроза, может потребоваться пересадка печени. Пересадка печени избавляет от заболевания в большинстве случаев.
Профилактика
Предотвратить заболевание нельзя. Если есть родственник первой степени — родитель, брат, сестра — с болезнью Вильсона-Коновалова, поговорите с врачами об обследовании вас и других членов семьи на предмет заболевания. Врач может поставить диагноз и начать лечение до появления симптомов и осложнений.
Ранняя диагностика и лечение могут уменьшить или предотвратить повреждение органов.
Прогноз
Прогноз улучшается при назначении адекватной терапии на ранних стадиях заболевания. Терапия на поздней стадии существенно не влияет на развитие и прогрессирование осложнений и на прогноз заболевания. Смерть наступает преимущественно в молодом возрасте, как правило, от осложнений цирроза печени (кровотечение из варикозно расширенных вен пищевода и желудка, печеночная недостаточность, фульминантная печеночно-клеточная недостаточность), реже — от осложнений, связанных с поражением нервной системы.
Медные люди: что важно знать о болезни Вильсона-Коновалова
Откуда берется медь в нашем организме и зачем она нужна? Из-за чего возникает болезнь, при которой медь не выводится из тела самостоятельно, и почему она опасна? Как живут люди, у которых меди слишком много, и чем медицина может им помочь?
Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы запускаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними. Читайте в ноябре рассказ о болезни Вильсона-Коновалова, которая встречается у одного из 30 тысяч человек, а также историю «медного пациента» — 38-летней Олеси Фрерихс, которая узнала о заболевании на 7 месяце беременности.
- Вероятность встретить редкого пациента у обычного врача ничтожно мала — в России редким (орфанным)считаетсязаболевание, которое обнаруживается у одного из 10 тысяч человек. В академических аудиториях им не уделяют должного внимания, а в повседневной профессиональной практике это приводит к диагностическим ошибкам, упущенному времени и поломанным судьбам.
- Информационно-просветительский гуманитарный проект «12 месяцев» — цикл материалов о редких (орфанных) заболеваниях и жизни людей с ними — реализуют студенты и ординаторы кафедры патологической анатомии Северо-Западного государственного медицинского университета имени И. И. Мечникова (Санкт-Петербург). Они изучают генетические методы диагностики и лечения, учатся разбираться в сложных вопросах медицины и рассказывать о них, находить общий язык с редкими пациентами и их родителями, работать в мультидисциплинарной команде.
Как связаны медь, гены и болезнь Вильсона-Коновалова?
Ежедневно наш организм получает с пищей не только белки, жиры, углеводы и витамины — в него поступает множество ионов металлов и других микроэлементов, необходимых для его правильного функционирования. Например, медь, которая содержится в печени и мясе, какао и бобовых, злаках и орехах.
Несмотря на небольшую суточную потребность — всего 1,5-2,5 мг, медь участвует в обмене энергии, метаболизме железа, защите клеточных мембран — то есть практически во всех физиологических процессах в нашем теле.
Обмен меди, как и многие другие индивидуальные особенности организма генетически запрограммированы, то есть заложены в нас при рождении. Гены — это всего лишь инструкции для синтеза белков в рибосомах. Один ген — один белок, все достаточно просто. А вот какую функцию будет выполнять этот белок — зависит от его структуры.
Некоторые белки участвуют в метаболизме меди. У всех людей всасывание меди происходит в желудке и двенадцатиперстной кишке. Дальше медь транспортируется в печень, где соединяется с различными белками, затем ее часть выводится в связанном состоянии в кровь и уже после — в мочу и кал.
Чтобы меди в организме было всегда ровно столько, сколько нужно, метаболизм регулирует и выравнивает скорости ее поступления и выведения наружу.
Ключевую роль в этом уравнении играет белок-транспортер меди под названием ATP7. Он работает исправно, если в его гене, в инструкции по его сборке, нет опечаток — или мутаций, как их называют биологи. Известно уже более 800 таких опечаток в гене АТР7.
Именно из-за этих мутаций у некоторых людей белок ATP7 не работает вовсе или его функция заметно снижена. В этом случае излишки меди не выводятся из организма, а накапливаются в органах. Но много — не всегда значит хорошо. Избыток металла не дает пациентам с неработающим белком ATP7 повышенную крепкость организма, а напротив — повреждает клеточные структуры. От избытка меди прежде всего страдает головной мозг и печень.
Такую болезнь, вызванную накоплением меди в организме из-за мутации белка ATP7, называют болезнью Вильсона — по имени Сэмюэля Уилсона (Вильсона) — британского невролога, подробно описавшего симптомы заболевания в 1912 году.
В России более распространено другое название — болезнь Вильсона-Коновалова: в 1960 году советский невропатолог Николай Коновалов существенно расширил понимание болезни. Еще реже можно встретить тройное название — Вильсона-Вестфаля-Коновалова (немецкий патолог Карл Фридрих Вестфаль описал болезнь еще в 1883 году). Сами пациенты называют себя вильсонятами.
В этом ролике — краткая история открытий на пути к познанию природы этой болезни и способов помочь пациентам.
Когда появляются симптомы и какими они могут быть?
Первые симптомы болезни Вильсона-Коновалова обычно появляются на втором или третьем десятке лет жизни в виде неврологических нарушений, например, нечеткости речи, нарушении глотания, автоматических жевательных движений, и нарушений показателей работы печени.
Болезнь умеет маскироваться: она может начаться как диспепсия (боль в верхнем отделе живота) или нарушение двигательной функции и речи. И даже самый запоминающийся ее признак — медное кольцо вокруг роговицы можно обнаружить только у половины больных.
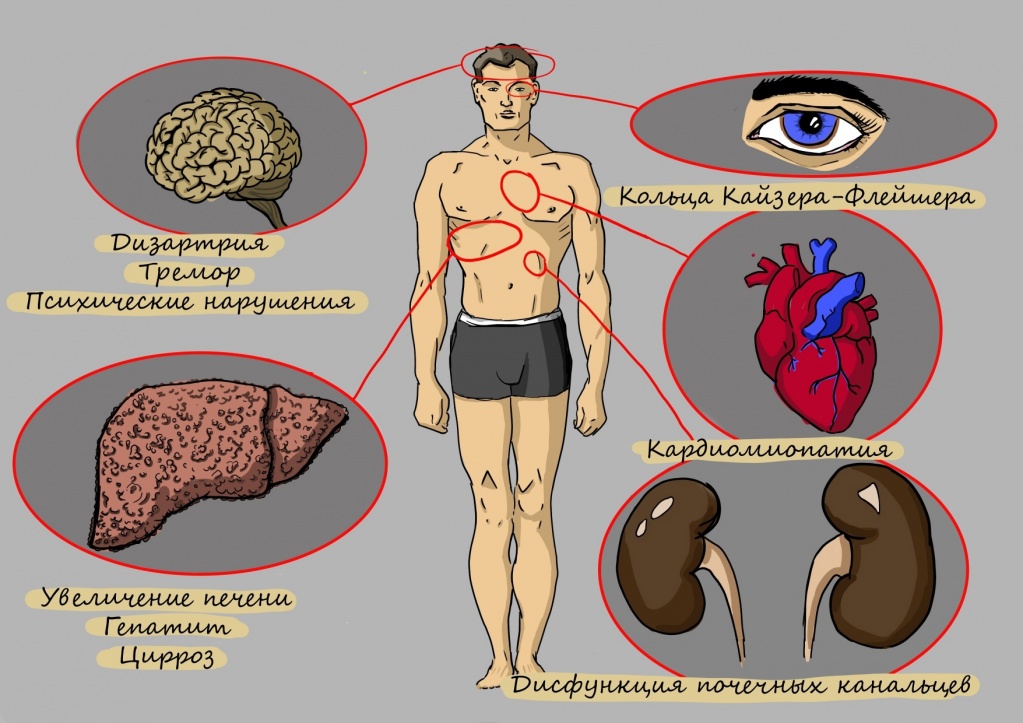
В теле не остается ни одного органа, равнодушного к нарушению обмена меди.
Но точная диагностика этого заболевания обычно представляет собой большую проблему. Биохимическое исследование показателей обмена меди в крови считается информативным, однако достоверно подтвердить мутацию можно только при помощи генетического анализа.
Чем медицина может помочь «медным людям»?
Главный на сегодняшний день способ лечения болезни Вильсона-Коновалова был предложен еще больше полувека назад — это регулярное применение препаратов, которые связывают медь и выводят ее из организма. Важным для «вильсонят» остается соблюдение строгой диеты и, при необходимости, пересадка печени.
Прерывание терапии или неправильное лечение может привести к смерти в течение нескольких месяцев. При этом медикаментозная, лекарственная терапия эффективна не для всех пациентов — побочные эффекты препаратов могут даже утяжелять состояние некоторых людей. Поэтому наука продолжает искать ответ — чем же помочь «вильсонятам»?
Не так давно исследователи научились исправлять «опечатки» в генах. В том числе — и в ATP7. Не всегда это получается хорошо, тем не менее, генная терапия может навсегда избавить пациентов с болезнью Вильсона-Коновалова от пожизненного приема препаратов и строгой диеты.
Как исследователи пытались научиться делать такую «операцию» на генах? Первые способы замены поврежденного участка гена на здоровый, в частности, применение олигонуклеотидов — коротких фрагментов ДНК или РНК, получаемых путем химического синтеза, даже у мышей приводили к едва заметному улучшению — его точно не хватило бы для помощи человеку.
Разочарования исследователей продлились до тех пор, пока они не научились «протезировать» больной ген. Для этого копию гена без поломки помещают в структуру искусственного вируса, чтобы эта неизмененная копия гена легко проникла в клетку. Освободившись от белковой оболочки, как от скафандра, ген начинает работать — и клетка наполняется правильно работающими переносчиками для меди.
Такая генная терапия представляет собой перспективную альтернативу классическому лекарственному лечению.
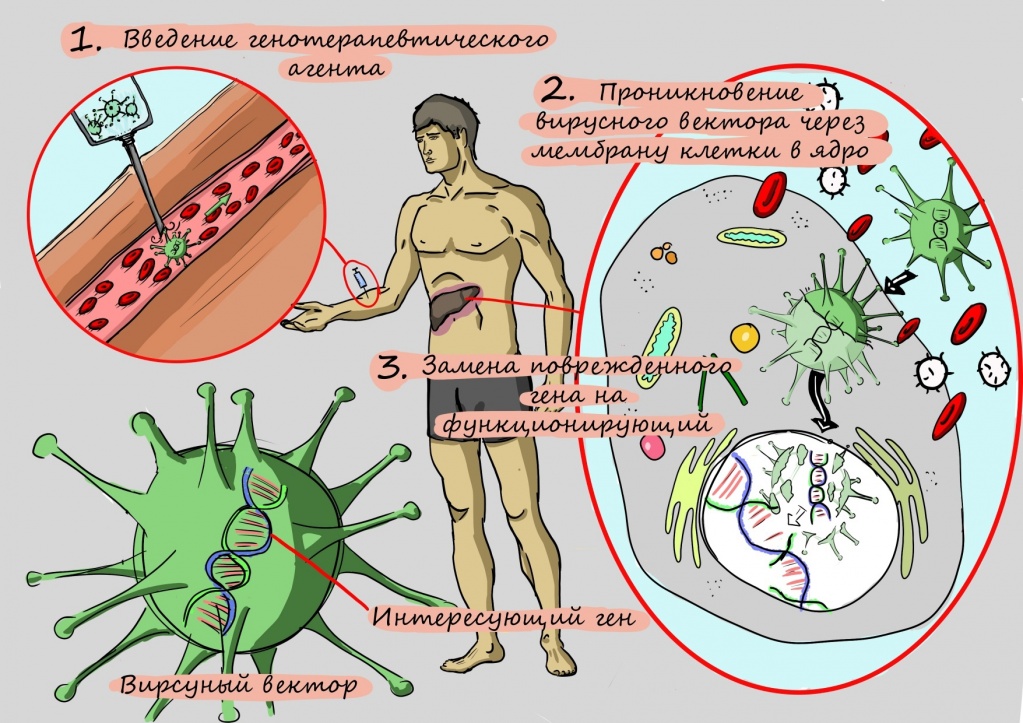
Молекулярное протезирование гена АТР7 — генная терапия
Однако при проведении этого вида терапии исследователи столкнулись с проблемой: аденовирус, который использовали для доставки гена в клетку, не может внедряться в собственный генетический аппарат клеток, — и это приводило к тому, что терапия работала недолго.
Следующим шагом в разработке терапии стало применение другого носителя — так называемого аденоассоциированного вирусного вектора.
Мышам с мутацией, которая похожа на мутацию в белке ATP7, вызывающую болезнь Вильсона-Коновалова, вводили такой вектор, и эффект оказался положительным.
Аденоассоциированный вирусный вектор имеет способность встраиваться в генетический аппарат клеток, поэтому результат лечения был более долгосрочным. Однако и этого оказалось недостаточно.
Успешный опыт подогревал ажиотаж исследователей: так, в 2021 году начались первые два клинических исследования по спасению «вильсонят» методами генной терапии (NCT04884815, NCT04537377). Через несколько лет мы узнаем — помогут ли эти разработки пациентам.
Материал подготовили: Роман Деев, Максим Пушкин, Екатерина Пичугина, Алексей Паевский, Виктория Рыжкова.
Иллюстрации Владислава Ефремова.
Болезнь Вильсона ( Болезнь Вестфаля-Вильсона-Коновалова , Болезнь Вильсона-Коновалова , Гепатолентикулярная дистрофия , Гепатоцеребральная дистрофия , Лентикулярная прогрессирующая дегенерация )
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона может протекать в брюшной, ригидно-аритмогиперкинетической, дрожательной или экстрапирамидно-корковой форме. Диагностика болезни Вильсона включает офтальмологическое обследование, биохимические анализы мочи и крови, МРТ или КТ головного мозга. Основу патогенетической терапии составляют тиоловые препараты, которые могут приниматься в течении нескольких лет и даже пожизненно.
МКБ-10
Общие сведения
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФ-азы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Первооткрыватель заболевания — А.К. Вильсон, описавший заболевание в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году. Понятию «болезнь Вильсона» соответствуют также: болезнь Вильсона-Коновалова, болезнь Вестфаля-Вильсона-Коновалова, дистрофия гепатоцеребральная, дистрофия гепатолентикулярная, дегенерация лентикулярная прогрессирующая.
Причины
Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3-q21.1). Организм человека содержит около 50-100 мг меди. Суточная потребность меди для человека — 1-2 мг. 95% абсорбированной в кишечнике меди, транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5% в форме комплекса с альбумином. Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохром-С-оксидаза и др.). При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтез главного медьсвязывающего белка (церулоплазмина) и выведение меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению.
Классификация
Согласно классификации Н.В. Коновалова различают пять форм болезни Вильсона:
- брюшная
- ригидно-аритмогиперкинетическая
- дрожательно-ригидная
- дрожательная
- экстрапирамидно-корковая
Симптомы
Для болезни Вильсона характерен клинический полиморфизм. Первые проявления заболевания могут появиться в детстве, юношестве, в зрелом возрасте и гораздо реже в зрелом возрасте. В 40-50% случаев Болезнь Вильсона манифестирует с поражения печени, в остальных — с психических и неврологических расстройств. С вовлечением в патологический процесс нервной системы обнаруживается кольцо Кайзера-Флейшера.
Брюшная форма развивается преимущественно до 40 лет. Характерный признак — тяжелое поражение печени по типу цирроза печени, хронического гепатита, фульминантного гепатита.
Ригидно-аритмогиперкинетическая форма манифестирует в детском возрасте. Начальные проявления — мышечная ригидность, амимия, смазанность речи, трудности при выполнении мелких движений, умеренное снижение интеллекта. Для этой формы заболевания характерно прогрессирующее течение, с наличием эпизодов обострения и ремиссии.
Дрожательная форма возникает в возрасте от 10 до 30 лет. Преобладающим симптомом является тремор. Кроме того, могут наблюдаться брадикинезия, брадилалия, тяжелый психоорганический синдром, эпилептические приступы.
Экстрапирамидно-корковая форма встречается весьма редко. Ее начало схоже с началом какой-либо из вышеперечисленных форм. Для нее характерны эпилептические припадки, экстрапирамидные и пирамидные нарушения и выраженный интеллектуальный дефицит.
Диагностика
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера. Биохимические исследования мочи обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца. Основным дифференциально-диагностическим признаком этих заболеваний является отсутствие характерных для болезни Вильсона кольца Кайзера-Флейшера и расстройств обмена меди. Для подтверждения болезни Вильсона проводится генодиагностика.
Лечение болезни Вильсона
Основой патогенетического лечения является назначение тиоловых препаратов, в первую очередь — D-пеницилламина либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза). Лечение D-пеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — леводопы, карбидопы, тригексифенидила.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень). Немедикаментозное лечение состоит в назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.).
Прогноз и профилактика
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме. Постоянный прием тиоловых препаратов по схеме, назначенной врачом-специалистом, позволяет поддерживать профессиональную и социальную активность пациента.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.
Источник https://clinic-a-plus.ru/articles/gastroenterologiya/21502-bolezn-vilsona-konovalova.html
Источник https://media.nenaprasno.ru/articles/keysy/mednye-lyudi-chto-vazhno-znat-o-bolezni-vilsona-konovalova/
Источник https://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/wilsons

