Синдром Марфана
Синдром Марфана – это системное заболевание, передающееся по аутосомно-доминантному типу наследования, характеризующееся недоразвитием соединительнотканных волокон, вследствие возникновения структурных дефектов в коллагене. Наиболее часто при данной патологии поражаются органы зрения, опорно-двигательный аппарат, сердце и сосуды. Прогноз при этой болезни будет определяться выраженностью сопутствующих патологических изменений. Большинство пациентов не доживают до пятидесяти лет.
Впервые симптомы, характерные для синдрома Марфана, были описаны в 1875 году американскими врачами. Свое название данное заболевание получило в честь французского врача-педиатра А. Марфана, на протяжении нескольких лет изучавшего эту болезнь.
В настоящее время синдром Марфана встречается не часто. В среднем уровень его распространенности колеблется от 1 случая на 20 тысяч человек до 1 случая на 5 тысяч человек. При этом с ним одинаково часто сталкиваются как представители мужского, так и женского пола. Какой-либо расовой детерминированности также не прослеживается.

Суперфуды в косметике: сочные коктейли для здоровья кожи и волос
Симптомы
Симптомы синдрома Марфана отличаются своим многообразием.
Пациенты, страдающие от данного заболевания, как правило, имеют специфический внешний вид. Они отличаются высоким ростом при условии относительно небольшой длины туловища и чрезмерно длинных верхних и нижних конечностей. Помимо того, что руки и ноги больного человека непропорционально длинные, они еще и очень тонкие. Отмечаются удлинение и истончение пальцев, слабое развитие подкожно-жировой клетчатки, снижение мышечного тонуса.
Голова пациента имеет длинную и узкую форму. Нередко встречается аномалия развития верхней челюсти, представленная выгибанием твердого неба вверх с образованием высокого свода. В результате челюстно-лицевой деформации наблюдается неправильный прикус.
Суставы больного человека избыточно подвижны (гипермобильны), грудная клетка деформирована с выступанием вперед (килевидная форма) или западением во внутрь (воронкообразная форма) грудины. Кроме этого, характерны различные деформации со стороны позвоночного столба, например, его искривление в передне-задней плоскости.
Типичным признаком синдрома Марфана являются различные патологические изменения со стороны сердечно-сосудистой системы. Они могут быть представлены дефектами сосудистых стенок, например, аневризмами, пороками развития клапанов и перегородок сердца, например, пролапсом митрального клапана, расширением корня аорты, и так далее. В 2013 году ученые из Национального медицинского исследовательского центра сердечно-сосудистой хирургии им. А.Н. Бакулева опубликовали результаты работы, в которой было установлено, что расширение корня аорты встречается у 60% пациентов с синдромом Марфана, пролапс митрального клапана – у 91%.
Кроме этого, при данном заболевании зачастую поражаются органы зрения. С клинической точки зрения это может проявляться миопией различной степени выраженности, эктопией хрусталика, косоглазием, патологическими изменениями со стороны роговицы и так далее.
Помимо вышеперечисленного, синдром Марфана может сопровождаться поражением и других органов, например, нервной или дыхательной системы, кожи и многого другого.
Формы
Формы синдром Марфана могут быть следующие:
Стертая форма — слабо выраженные патологические изменения обнаруживаются в одной или двух системах организма.
Выраженная форме – могут отмечаться слабо выраженные симптомы со стороны трех и более систем, выраженные симптомы со стороны одной, двух и более систем.
Кроме этого, данное заболевание может иметь прогрессирующее или стабильное течение.
Причины
Причина синдрома Марфана – это генетическая мутация, передающаяся по аутосомно-доминантному типу наследования.
При этом ведущая роль в возникновении данной патологии отводится мутации в гене FBN1, отвечающем за выработку специфического гликопротеида – фибриллина. В результате нарушения синтеза фибриллина изменяется структура коллагеновых волокон, появляются проблемы с образованием волокнистых структур, соединительная ткань теряет свою прочность и упругость, не может выдерживать физиологические нагрузки.
Примерно в семидесяти пяти процентах случаев у человека с данным диагнозом выявляются родственники, страдающие от аналогичной проблемы. Значительно реже мутация в гене FBN1 является первичной. Замечено, что вероятность рождения ребенка с данной болезнью находится в прямой зависимости от возраста мужчины. Чем старше отец, тем выше риски.
Методы диагностики
Диагностика синдрома Марфана начинается с объективного осмотра, сбора анамнеза.
В настоящее время разработаны диагностические критерии данного заболевания:
Со стороны опорно-двигательного аппарата большими критериями считаются наличие килевидной или воронкообразной формы грудной клетки, искривление позвоночника во фронтальной плоскости более двадцати градусов, смещение вышележащего позвонка относительно нижележащего, плоскостопие, смещение медиальной стенки вертлужной впадины в сторону полости таза и так далее.
Со стороны сердечно-сосудистой системы к большим критериям относятся расширение корня аорты или расслоение ее восходящей части.
Со стороны органов зрения единственным большим критерием является эктопия хрусталика разной степени выраженности, выявляемая в 50-80% случаев.
Со стороны нервной системы единственный большой критерий – это эктазия твердой мозговой оболочки.
Все остальные патологические изменения со стороны внутренних органов принято относить к малым критериям. Постановка диагноза «синдром Марфана» основывается на наличии как минимум одного большого критерия со стороны двух систем и одного малого критерия со стороны третьей системы. Кроме этого, диагноз считается подтвержденным при наличии четырех и более больших диагностических критериев со стороны опорно-двигательной системы.
Из инструментальных методов исследования при данном заболевании назначаются:
- Рентгенография грудной клетки и тазобедренных суставов.
- Электрокардиография.
- Эхокардиография.
- Аортография.
- Компьютерная и магнитно-резонансная томография сердца и сосудов.
- Офтальмоскопия, а также многое другое.
Для окончательного подтверждения диагноза могут прибегать к помощи генетического исследования.
Лечение
Лечение синдрома Марфана направлено на то, чтобы остановить прогрессирование болезни и не допустить развития осложнений, в особенности, со стороны сердечно-сосудистой системы.
Из лекарственных препаратов для купирования симптомов, указывающих на поражение сердца и сосудов, могут назначаться бета-адреноблокаторы, ингибиторы ангиотензинпревращающего фермента, антагонисты кальция. Недостаточность сердечных клапанов, аневризма грудного отдела аорты диаметром пять и более сантиметров, расслоение аорты – все это является показанием к хирургическому вмешательству.
Особого внимания требуют беременные женщины с наличием данной патологии. В 2016 году ученые из Омского государственного медицинского университета опубликовали работу, по результатам которой было установлено, что при синдроме Марфана частота расслаивающей аневризмы аорты во время беременности составляет 4,5-6% случаев. При расслоении аорты в первом и втором триместре беременности необходимо провести экстренное кардиологическое хирургическое вмешательство, в третьем – сначала оперативное родоразрешение, а только затем протезирование аорты.
В остальном тактика лечения также будет определяться имеющимися патологическими изменениями. Так, например, при наличии катаракты требуется ее удаление при помощи хирургического или лазерного вмешательства, при деформации грудной клетки – нужна торакопластика.
Профилактика
Профилактика синдрома Марфана не разработана.
Какие вопросы следует задать врачу
Что такое синдром Марфана?
Из-за чего развивается данное заболевание?
Какие симптомы характерны для синдрома Марфана?
Как можно подтвердить диагноз?
Какова тактика лечения при выявлении синдрома Марфана?
Советы пациенту
Поскольку какие-либо способы предупредить развитие синдрома Марфана не разработаны, парам, собирающимся стать родителями, рекомендуется на этапе планирования беременности посетить врача-генетика для консультации и выявления возможных рисков.
Синдром Марфана

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
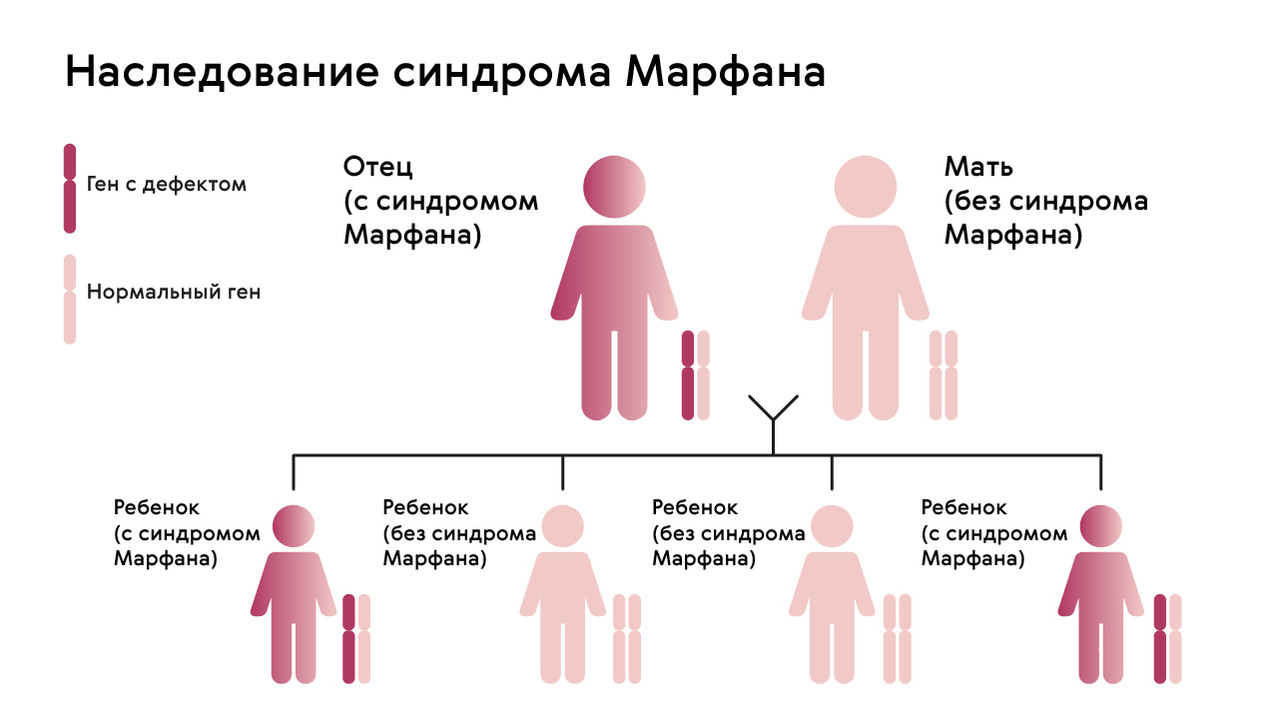
Рисунок. 1. Схема наследования синдрома Марфана. Источник: МедПортал
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5–10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
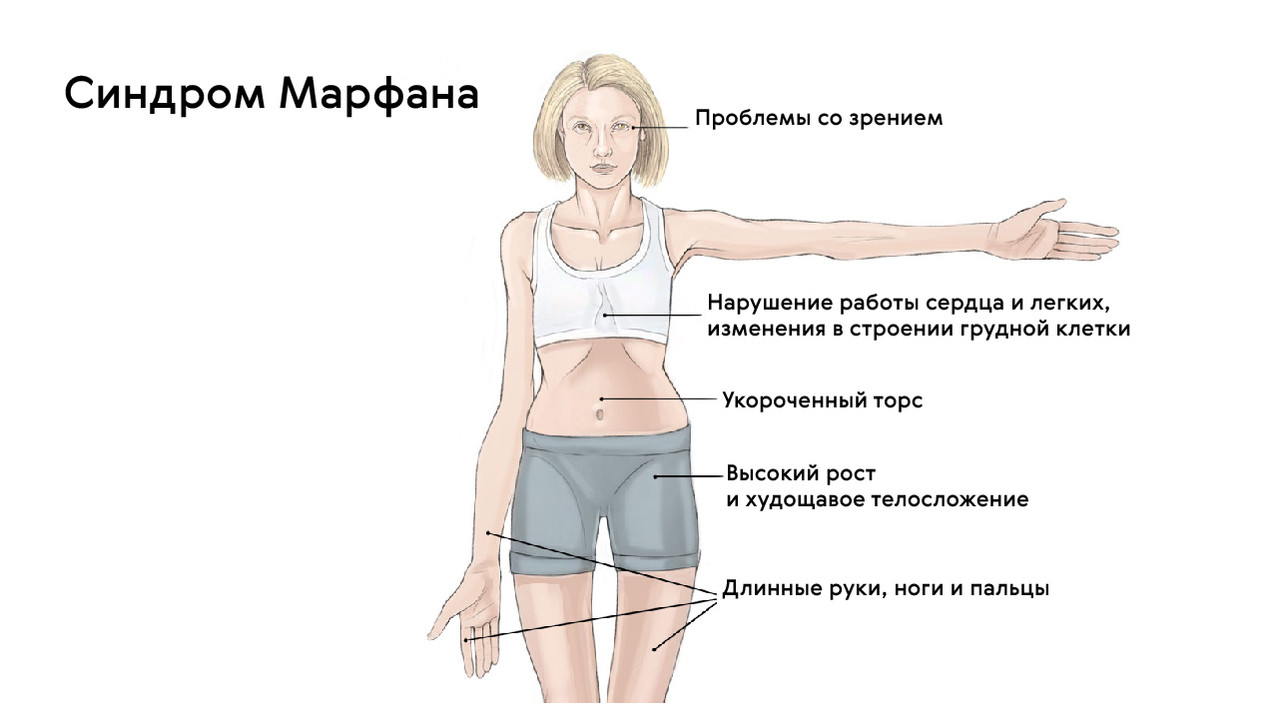
Рисунок 2. Классические проявления синдрома Марфана. Источник: zinkmd.com
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
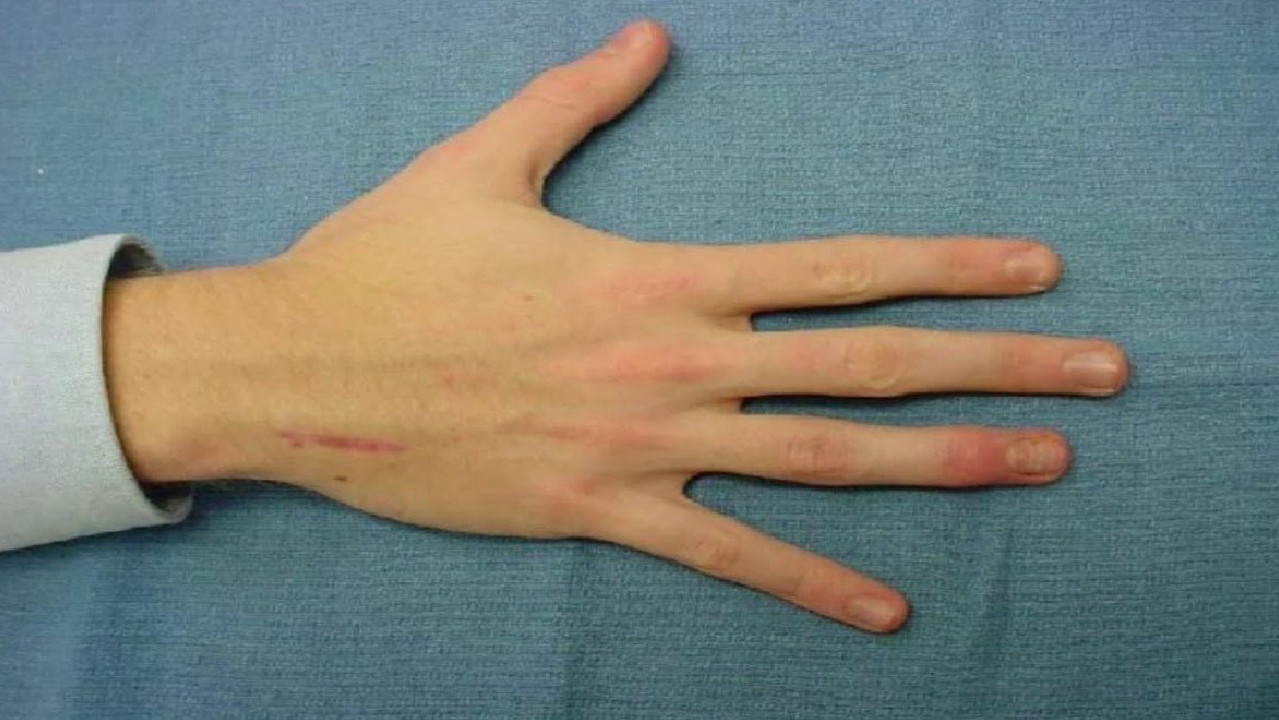
Рисунок 3. Арахнодактилия. Источник: twitter.com
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
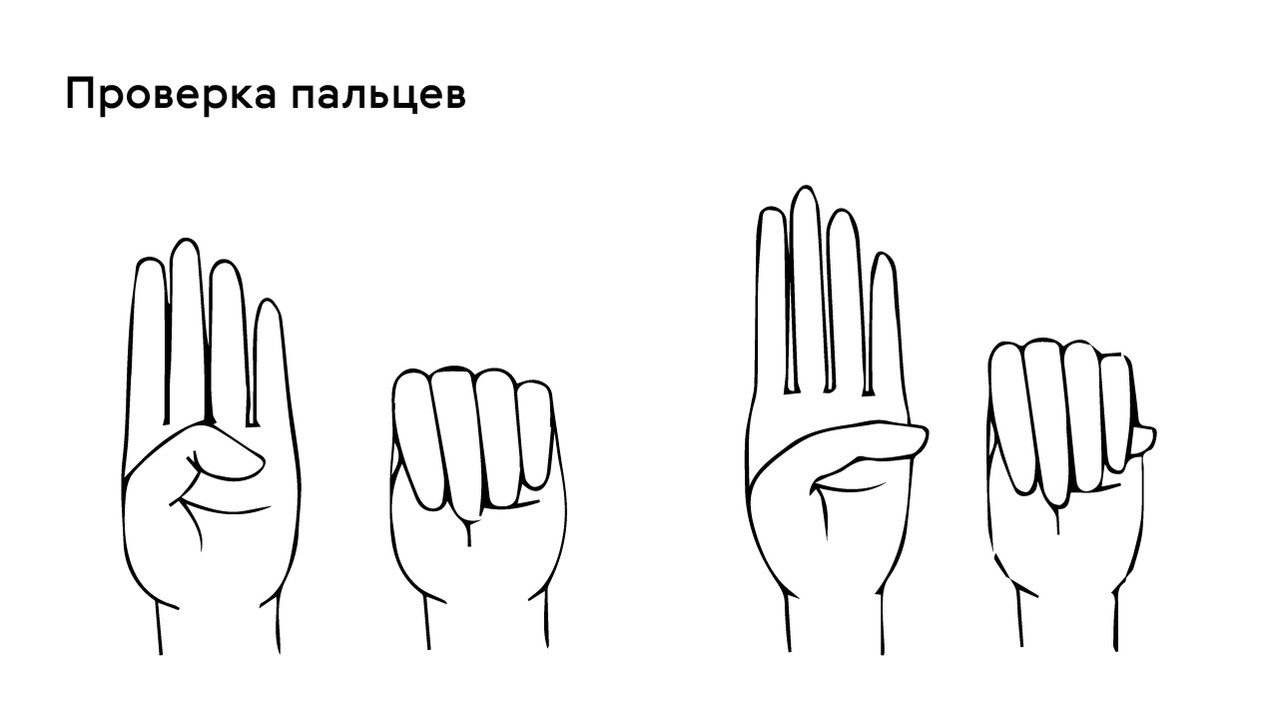
Рисунок 4. Проверка на арахнодактилию. Источник
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
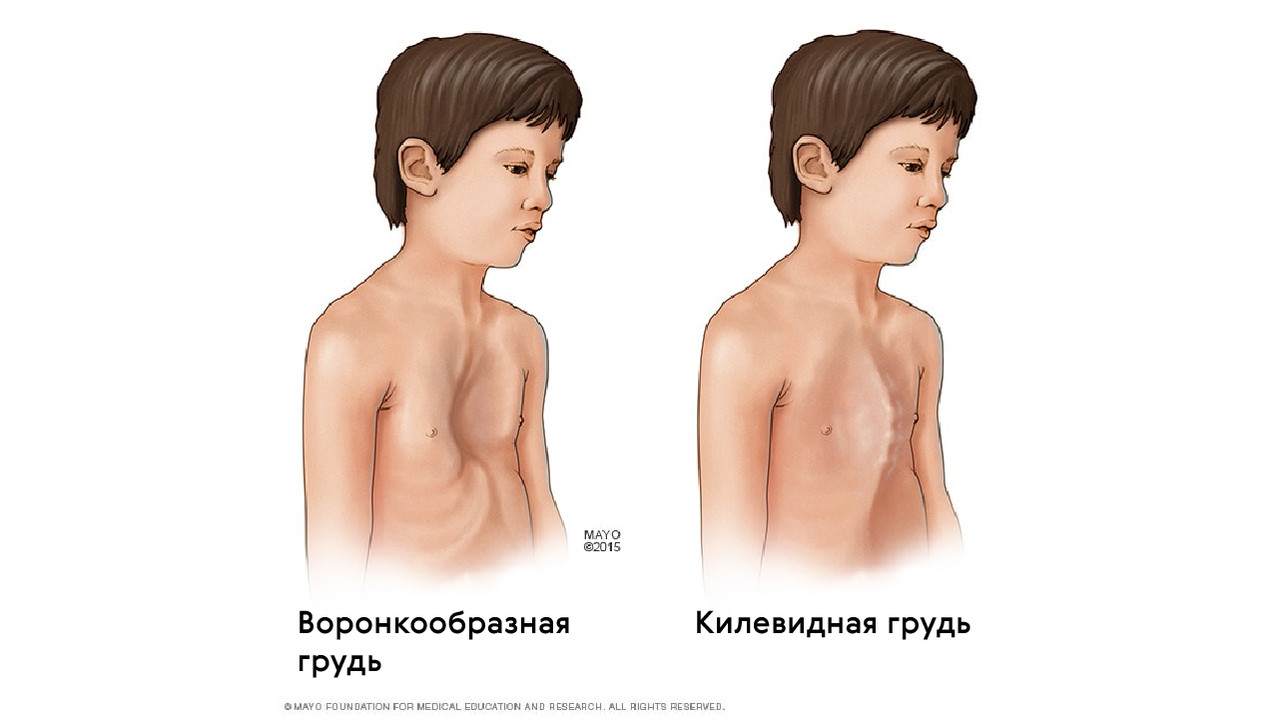
Рисунок 5. Воронкообразная и килевидная деформации грудной клетки при синдроме Марфана. Источник: mayoclinic.org
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65–100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).

Рисунок 6. Актер Хавьер Ботет, страдающий синдромом Марфана. Источник: kinopoisk.ru
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
«Мое сердце пережило уже три операции»: что нужно знать о синдроме Марфана и жизни с ним
Что может объединять датского писателя Ганса Христиана Андерсена, советского поэта Корнея Чуковского, президента США Авраама Линкольна, французского президента Шарля де Голля, итальянского скрипача Никколо Паганини, русского пианиста Сергея Рахманинова и американскую чемпионку Олимпийских игр Фло Хайман?
Считается, что, помимо безусловного таланта, их также связывало одно из самых распространенных наследственных заболеваний соединительной ткани — синдром Марфана. Высокий рост, длинные конечности, вытянутые и тонкие пальцы рук, особенности строения грудной клетки и подвижные суставы — вот основные внешние признаки этой болезни. Главная ее угроза — разрыв самой крупной артерии нашего организма — аорты.
Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы продолжаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними. Шестая часть — рассказ о синдроме Марфана, с которым рождается один из 5000 тысяч детей.
Почему возникает и как проявляется синдром Марфана
Синдром (или болезнь) Марфана — генетическое заболевание, то есть в его основе лежит «поломка» в строении и функции гена. Оно передается по наследству по аутосомно-доминантному типу: как правило, если синдром есть у одного из родителей, то с вероятностью 50% болезнь будет и у ребенка. Только в 27% случаев возникает спонтанная генетическая мутация, и у здоровых родителей рождается ребенок с синдромом Марфана.
Комплекс симптомов, характерных для этого синдрома, был впервые описан французским педиатром Антуаном Бернардом Марфаном. Клинический случай своей 5 летней пациентки по имени Габриель он представил на заседании медицинского общества в Париже в 1896 году. Хотя сегодня считается, что у Габриель было совершенно другое заболевание, ее внешние признаки навсегда получили фамилию врача, впервые объединившего их в отдельный синдром.
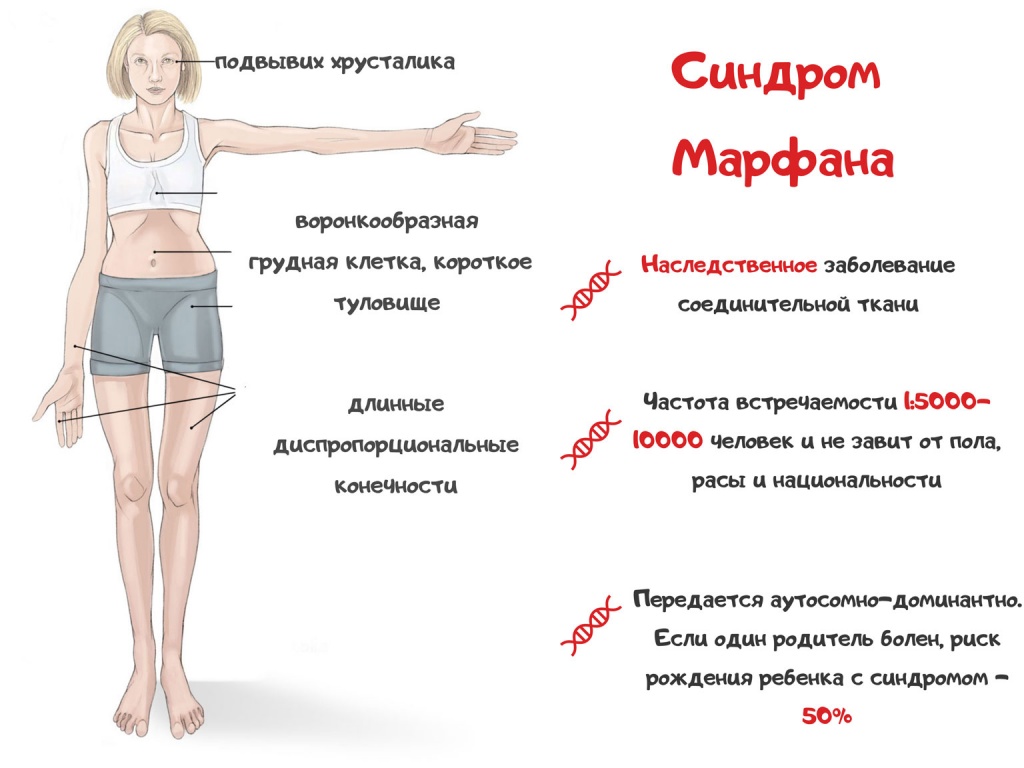
Внешние признаки синдрома
Молекулярная основа заболевания — «поломка» гена — была найдена в 1991 двумя научными коллективами под руководством Брендана Ли и Гарри Дитца.
Причина заболевания кроется в мутации в гене FBN1, который кодирует белок соединительной ткани — фибриллин-1. Этот белок — компонент микроволокон (микрофибрилл), которые обеспечивают структурную поддержку эластичной и неэластичной соединительной ткани по всему телу. Например, такие волокна в большом количестве присутствуют в кровеносных сосудах, в том числе, в стенке аорты, клапанах сердца, легких, связках.
В результате «поломки» в генах нарушается структура фибриллина-1, и он больше не может формировать прочный каркас эластических волокон. Из-за этого их количество уменьшается на 15-20%, они теряют свои свойства, подвергаются перерастяжению, что приводит к самому серьезному осложнению синдрома Марфана — формированию аневризмы (расширения определенного участка) аорты и ее последующему разрыву, способному привести к смерти пациента.
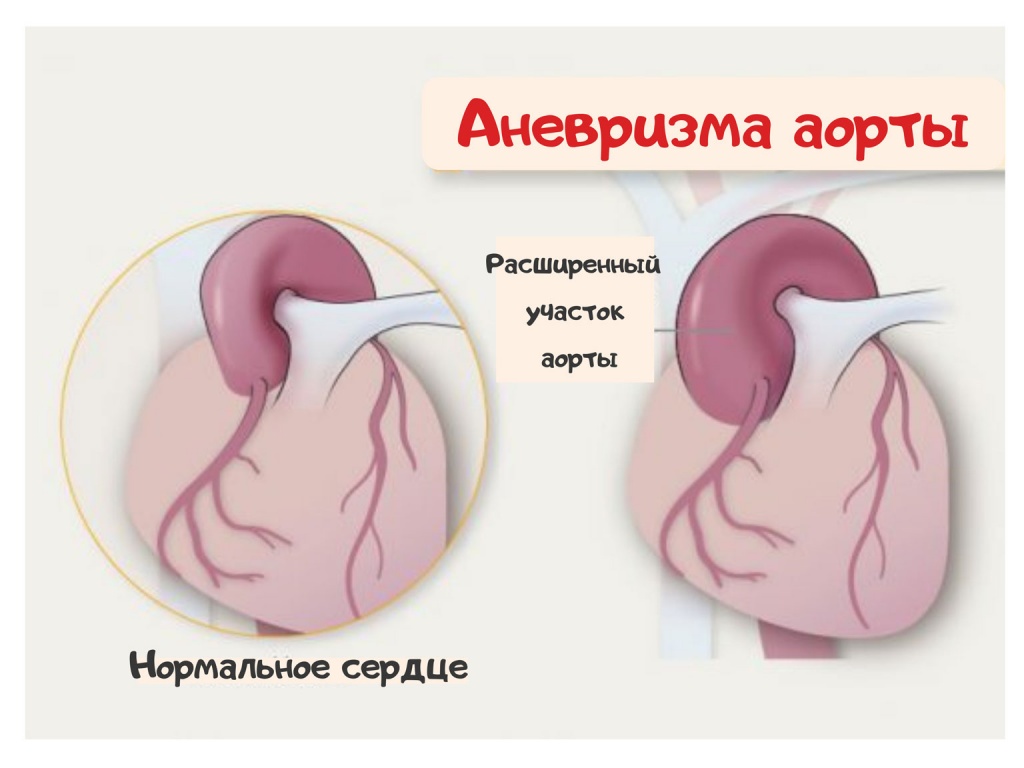
Аневризма аорты. Осложнение синдрома Марфана, которое может привести к смерти пациента.
Кроме этого, часто поражается митральный клапан: митральная недостаточность проявляется одышкой, быстрой утомляемостью и учащенным сердцебиением.
Из-за повсеместного распространения соединительной ткани в организме, а, следовательно, и белка фибриллина-1, страдает не только сердечно-сосудистая система, но также и опорно-двигательная, зрительная и дыхательная.
Синдром Марфана очень разнороден: описаны как легкое течение заболевания, так и крайне тяжелое. В первом случае его могут диагностировать в подростковом возрасте или даже на третьем десятке жизни человека, а самое тяжелое течение или неонатальная форма синдрома проявляются с рождения и средняя продолжительность жизни таких пациентов составляет приблизительно 4 года.
Вариативность клинических проявлений обусловлена характером изменений в гене фибриллина-1. Например, легкое или среднее течение заболевания встречается при потери одной копии гена, что приводит к недостаточному синтезу (количеству) нормального, хорошего фибриллина-1, а при неонатальной, самой тяжелой форме, мутации чаще всего происходят в конкретных местах гена — в 24-32 кодирующих участках (экзонах).
Сегодня средняя продолжительность жизни пациентов с синдромом Марфана (кроме неонатальной формы) составляет 70 лет, хотя еще в 70-х годах прошлого столетия 50 % мужчин умирали к 40 годам, а 50% женщин — к 48 годам. Основная причина смерти — развитие осложнений со стороны сердечно-сосудистой системы.
Как диагностировать синдром Марфана
Как правило, постановка диагноза не вызывает трудностей, так как синдром Марфана имеет яркую триаду признаков: высокий рост и длинные конечности, эктопию хрусталика (подвывих, смещение вверх) и аневризму аорты.
Основной инструмент диагностики — это тщательный осмотр, сбор анамнеза, инструментальное и клиническое обследование несколькими специалистами: генетиком, кардиологом и офтальмологом.
Дифференциальную (то есть сравнительную) диагностику проводят с синдромом Лойса-Дитца и MASS-фенотипом, признаки которых похожи на синдром Марфана. Диагноз выставляется на основании Генстких критериев и балльной оценке системного вовлечения соединительной ткани в патологический процесс.
Выделяют два самых важных критерия — расслоение аорты и подвывих хрусталика. Остальным клиническим признакам присваивается от 1 до 3 баллов. Например, наличие пролапса митрального клапана — это 1 балл, а признак запястья и большого пальца (когда верхняя, дистальная фаланга большого пальца выступает за край сжатого кулака) — 3 балла.
Еще один важный диагностический маркер — Z-критерий, который отражает увеличение размера корня аорты.
Существуют онлайн-калькуляторы и опросники, которые помогают врачам и пациентам установить правильный диагноз. В перспективе для оценки риска расслоения аорты может быть использован анализ крови на специальные молекулы — циркулирующие микроРНК, прежде всего. Так, у пациентов с аневризмой аорты присутствует miR-574-5p.
Есть ли лечение при синдроме Марфана
Синдром Марфана — пока неизлечимое заболевание, и основные усилия врачей направлены на предотвращение серьезных сердечно-сосудистых осложнений. Для этого применяют хирургическое лечение в виде протезирования клапанов и операции по реконструкции аорты, а также медикаментозную терапию.
Еще в 1994 году были проведены клинические исследования, показывающие эффективность применения бета-блокаторов для замедления процессов расширения корня аорты. Однако сообщество Кокрейн (международная некоммерческая организация, объединяющая специалистов в области доказательной медицины — прим.ред.) говорит о низкой доказательности проведенных работ и о том, что статистически значимое снижение расширения корня аорты еще не свидетельствует о клинической пользе — то есть предотвращении разрыва аорты и снижения смертности.
Другая группа применяемых препаратов — блокаторы рецепторов ангиотензина. Обе этих группы могут быть вам известны как препараты «для снижения давления»: пациентам с синдромом Марфана они действительно помогают снижать нагрузку на сердце и аорту.
- Комбинация бета-блокаторов и блокаторов рецепторов ангиотензина сегодня считается наиболее эффективной тактикоймедикаментозной терапии.
Исследование этой технологии проводилось в 2018 году на клеточных линиях (клетки определенного типа, которых «выращивают» в пробирке) и человеческих эмбрионах. Они искусственно получали мутацию, характерную для синдрома Марфана, а затем с помощью геномного редактирования исследователи пытались эту ошибку исправить. Эффективность «исправления поломки» без возникновения нежелательных эффектов в среднем составила 89%
Основная сложность разработки генной терапии при синдроме Марфана — необходимость исправить весь объем соединительной ткани. Поэтому ведутся не только разработки препаратов, способных полностью излечить от этого заболевания, но и поиски молекулярных мишеней, которые смогут значительно улучшить качество жизни пациентов и предотвратить серьезные осложнения со стороны сердца и сосудов.
Исследования на мышах показали, что ингибирование (подавление) некоторых ферментов предотвращает сосудистую дисфункцию и может быть эффективной профилактикой развития аневризмы аорты.
Где могут помочь людям с синдромом Марфана
Несмотря на то, что заболевание пока остается неизлечимым, люди с синдромом Марфана могут жить полноценной, насыщенной жизнью, заниматься умеренной физической нагрузкой и иметь детей, как героиня нашего интервью (оно ниже).
- Есть сообщество «Marfan Foundation», где собрана актуальная информация о синдроме, возможностях его лечения и адаптации жизни с заболеванием с риском разрыва аневризмы аорты.
«Без болезни я бы дольше шла к себе»: история Марии и ее жизни с синдромом Марфана
«Я иногда сама себе завидую. Смотрю на свои сторис и думаю: «Боже, это же моя жизнь!» — так говорит 32-летняя Мария, поэтесса, писательница, наездница, мама шестилетней Авроры. А еще человек с синдромом Марфана — редким генетическим заболеванием, из-за которого Мария перенесла уже три операции на сердце.

«Я перестала скрывать свою болезнь, только когда выросла»
В моей семье никто не страдает синдромом Марфана: оба родителя не носители генетической мутации, старшие брат и сестра здоровы.
Одна из гипотез почему произошла мутация в моих генах — это облучение. Когда мама была беременной, она работала в зоопарке, и в этот период обезьяны начали болеть туберкулезом. Их возили на рентгенологическое обследование и во время процедуры держали на руках как детей — из-за этого мама и я получили большую дозу облучения.
В детстве мы с сестрой часто бывали в зоопарке: хоть мама уже там не работала, она приводила нас к своим подругам, чтобы мы им помогали и заодно проводили время на летних каникулах. Представляете, мы в 7-8 лет купали медведей! Думаю, что, если бы это все проходило в наше время, маму бы просто лишили родительских прав. Детство в зоопарке учит некоторым реалиям жизни, например: тебе иногда приходится быть жестоким, чтобы быть добродетельным.
В моем детстве и жизни в целом не было буллинга. Хотя у меня всегда косили глаза и торчала кость из груди (для синдрома Марфана характерно неправильное строение грудной клетки, она как будто выпирает, — прим. автора), это никогда не мешало мне в социальном плане. Я была популярной девочкой, среди лидеров класса, с друзьями и активной жизнью, на пятерки сдавала все нормативы по физкультуре и даже в какой-то период бегала на длинные дистанции быстрее всех одноклассниц, мне присылали валентинки…
Но мои родители скрывали от окружающих мое заболевание. Считалось, что никто не должен знать о болезни. Я понимаю маму: ей просто было страшно, что она не сможет меня защитить. Ей было важно, чтобы я была такая же, как все.
Только когда я выросла, я перестала скрывать свою болезнь. Принятие и признание обществу случилось уже после ЭКО, родов и первой операции на сердце. В какой-то момент я поняла, что у меня такое поддерживающее окружение, что просто малодушно что-то скрывать.
«Я не встречала некрасивых людей»
Когда мне было лет 16, я мечтала быть кинорежиссером и учиться во ВГИКе. Но мне говорили: «Ты чего, Маша? Девочек в таком возрасте туда не берут»! Тогда мама сказала: «Слушай, да не поступай никуда. Попутешествуешь, посмотришь на мир. А потом поймешь». И мы с друзьями поехали в летний лагерь, занимались там гребно-парусным спортом.
В то же лето мама купила нам с другом билеты в Питер, чтобы мы поехали поступать. Единственный ВУЗ, до которого я дошла, был Институт кино и телевидения. Я поступила на продюсера, но к 3 курсу поняла, что это совсем не мое, и ушла.
В тот момент решила, что очень хочу стать актрисой. Однако оказалось, что 20 лет — слишком поздно, чтобы поступать на актерский факультет в России. В итоге в тот год я училась сначала в Китае, потом в Аргентине (учила испанский), затем уехала в США и там поступила на классный актерский курс. А потом случилась моя первая операция…

Сейчас я зарабатываю тем, что пишу об искусстве. Моя главная задача — сделать текст интересным и живым. Чтобы человек, который пришел на выставку, понял ее смысл и проникся. Мне очень помогает моя работа — мои представления о красоте и мое видение мира изменились.
Сейчас я действительно скажу, что красота в глазах смотрящего. За много лет своей работы и психотерапии я не встречала некрасивых людей.
«Твой диагноз выставляется посмертно»
Мое сердце пережило уже три операции. Первая состоялась в 2011 году, мне было 22 года. В ноябре я сходила на плановую флюрографию, и врачи сказали: «У тебя очень огромное сердце, просто беги к кардиологу». Потом было УЗИ сердца, на нем тоже все «охали».
Мы с мамой узнали, что в клинике Шарите в Германии работает профессор Роланд Хетцер, который специализируется на реконструкции клапанов сердца, в том числе и пациентов с заболеваниями соединительной ткани. Операция и сопровождение в течение года стоили 50 тысяч евро. У мамы, конечно, не было таких денег — нам помогли ее друзья.
Запомнилось, что в Германии очень много разговаривают с пациентом: показывают картинки, презентации, обговаривают все детали операции, разъясняют все-все страховые случаи. Рассказали даже, что могут уронить с носилок! Впечатлительные и эмоциональные люди могут от этого впасть в панику. Я просто ждала, что мне скажут, что все будет хорошо.
В 2021 году была моя вторая операция на сердце — уже в Калининграде. В связи ковидными ограничениями было непонятно, сможем ли мы успеть в Германию.
В кардиоцентре оказалось очень хорошо, персонал заботливый и милый. В больнице суперчисто и аккуратно. Однако, по моему опыту, это, скорее, исключение, так как многим нашим врачам не хватает мягкости и эмпатичности, они не всегда думают, что говорят пациенту и какие эмоции он может из-за этого испытывать. Например, в России во время одного из обследований врач сказал мне: «Вообще я и часа жизни тебе не дам, твой диагноз выставляется посмертно». После этих слов у меня случилась первая паническая атака.
Самая страшная операция — третья. Она была 29 апреля 2021 года, всего год назад. У меня произошло расслоение аорты, и оказалось, что я проходила в таком состоянии три недели — просто какое-то чудо. После планового УЗИ меня госпитализировали в Центр Алмазова в Санкт-Петербурге и срочно прооперировали.
Эта операция оказалась и самой сложной в психологическом плане для мой дочери.
«Если бы не синдром, я бы вряд ли была сейчас мамой»
Плюс моего заболевания в том, что мне нужно быстро принимать решения. Так, в 22 года я решила, что буду мамой.
Профессор Хетцер перед первой операцией пришел ко мне в палату и сказал: «Я, конечно, надеюсь, что получится реставрировать твой клапан. Но если не получится сохранить собственный, то нам придется поставить искусственный: какой ты хочешь?» Говорю: «Я не знаю». И он ответил не по правилам: «Если бы ты была моей дочерью, я бы советовал тебе ставить биологический».
Наверное, считается, что таких фраз врачи не должны говорить, но… я всегда хотела ребенка, а выносить его можно только с биологическим клапаном, который нужно менять. До первой замены мне дали 5 — максимум 7 лет.
Еще до свадьбы мы с моим парнем начали ездить в Германию, в клинику, где собирались делать ЭКО. Всем эмбрионам перед подсадкой делали генетическое тестирование, чтобы подсадить только тех, у кого не было синдрома Марфана.
Рожать я тоже решила Германии: в одной из частных клиник в России моей маме прямо сказали, что она хочет избавиться от больной дочери, заведя здорового внука… Затем заведующая так оценила мою беременность: «Ну, 19 недель. Конечно, поздно с тобой уже об аборте говорить». Что? Я же пришла после ЭКО! Этот ребенок желанный!
Я листала все сборники имен, включая даже древних друидов, и выписывала из них понравившиеся, и первым именем в этом списке было Аврора. Ей уже 6 лет, и если бы не синдром, то я бы вряд ли была сейчас мамой.

«Жить, как хрустальная ваза, невозможно»
Моя жизнь удивительна, но в ней было несколько тяжелых периодов.
Первый случился, когда мне было 15 лет. Я перешла в физико-математическую школу, где дети были очень неэмоциональными — из-за этого мне было одиноко и плохо, я чувствовала себя инородным объектом.
Депрессия наступила после первой операции. Я просто попала в кромешную темень. Мне было очень сложно принять все манипуляции с моим телом. Плюс у меня были очень сильные головные боли, меня постоянно рвало. 9 дней мне кололи обезболивающие — сначала меня тошнило, но когда переставали вводить, возвращались головные боли. Был замкнутый круг, и мне казалось, это не кончится никогда.
Физически я вообще ничего не могла делать: чтобы встать с кровати, мне приходилось падать на пол и с четверенек подниматься. Кому ты в таком состоянии нужна? Какой ребенок? Какое будущее? Зачем я живу? Еще и шрам через все тело. Мне казалось, что меня в жизни никто никогда не полюбит, и было так плохо, что казалось: лучше умереть.
Из этого состояния мне очень помог выбраться мой бывший муж, друзья, родственники — люди, которые проявляли любовь. После операции я вернулась в Россию очень слабая, и все близкие бесконечно за мной ухаживали. Я только бровью поведу, а они уже бегут ко мне с чашкой чая.
Позже я уехала на реабилитацию в санаторий, а мама купила кучу продуктов и попросила моего бывшего мужа мне их привезти.
Помню, я сижу и плачу на диване в полной темноте, и он заходит ко мне в номер: «Слушай, я такой грязный и голодный, пойду в душ, а ты сделай мне, пожалуйста, чай с бутербродом». Такое простое, житейское отношение очень помогло мне в тот момент.
Бывший муж — единственный, кто не относился ко мне, как к хрустальной вазе. Жить, как хрустальная ваза, невозможно.
«Я благодарна своей болезни»
С 3-х лет я катаюсь на лошадях и даже не представляю каково это — не владеть верховой ездой. Лошади — это мой манифест свободы. На лошади я чувствую себя абсолютно здоровой и всемогущей, в отличие от ходьбы по лестнице: когда поднимаюсь на третий этаж, у меня начинается сильная одышка.

Я правда благодарна своей болезни: без нее я бы, наверное, дольше шла к себе и к тому, что делает меня счастливой. Сейчас мне настолько все равно на некоторые вещи, что, например, если выгляжу плохо, то думаю: «Зато выгляжу! У меня было три операции на сердце, а я еще ничего».
Да, я не знаю, когда у меня будет следующая операция и как она пройдет. Но на самом деле все люди живут в этой неизвестности и ожидании — я просто постоянно об этом помню. А еще я уверена, что ни одна энергия не появляется и не исчезает бесследно. Поэтому не могу поверить в конечность бытия.
Источник https://medaboutme.ru/zdorove/spravochnik/bolezni/sindrom_marfana/
Источник https://medportal.ru/enc/rheumatology/systemic/sindrom-marfana/
Источник https://media.nenaprasno.ru/articles/kolonki/chto-nuzhno-znat-o-sindrome-marfana-i-zhizni-s-nim/
